Advanced Bioimaging
Optical microscopes have been instrumental in the study of the life sciences for centuries. Since the invention of the laser, many advanced bioimaging techniques using microscopy have been created with progress accelerating greatly in the last two decades. Limitations in optical penetration depth, due to the scattering of light, originally limited studies to thin samples and, by necessity, processes taking place outside of the organism (ex vivo). In recent years, in vivo techniques have been developed that can visualize within the living organism or cell and have led to greatly increased understanding of cell function. Any bioimaging technique requires generating a signal from each portion of the cell or organism through some method of contrast. Some of these techniques require an exogenous contrast agent (originating outside of the organism or cell) to be added to the sample under investigation, such as a fluorescent dye. These agents can be specifically engineered and targeted to particular molecules. In other cases, the signal can be generated with endogenous contrast agents (those that originate naturally from within the organism or cell). This route has the advantage of not perturbing the original tissue micro-environment and limits possible cell toxicity.
Traditional microscopy techniques provide excellent spatial resolution, typically below 1 µm, in the lateral directions (within the plane of the sample). However, the axial resolution, into the depth of the sample, is typically much poorer. Multiple advanced bioimaging techniques have been developed to improve the axial resolution and allow a true 3D representation of the sample to be reconstructed. Chief among these techniques is confocal microscopy, where the axial resolution is improved by the addition of a confocal aperture that discriminates against light emanating from different depths in the sample. Confocal microscopy is described in Two-Photon Fluorescence Microscopy.
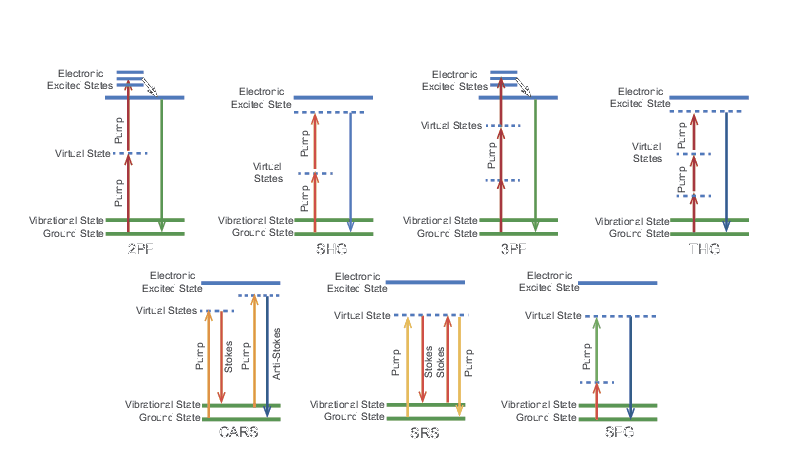
A wide range of laser-based nonlinear microscopies have been developed in the last two decades that make use of the high peak powers available from ps and fs laser sources. In each case, the excitation is due to an intensity-dependent effect which is most efficient at the focus of the laser beam. This can improve the axial resolution to about 1 µm without the use of a confocal aperture. Super-Resolution Microscopy, Two-Photon Fluorescence Microscopy, and Three-Photon Fluorescence Microscopy describe nonlinear microscopy techniques and a summary of the various mechanisms that enable these techniques is presented in Table 1. Laser Spectral Tunability describes various types of wavelength conversion and generation processes that are pertinent to these techniques. All of the techniques require a high peak power ultrafast laser and some require two synchronized ultrafast lasers at different wavelengths. Some techniques are applicable to endogenous contrast agents while others require the addition of a fluorescent dye. Figure 1 shows the energy level diagram for each mechanism which reveals how the laser light interacts with the system to produce a measurable signal. These diagrams will be referred to in the following sections.
| Technology | Fluorescent Dye | Pulse Duration | Number of Synchronized Wavelengths |
|---|---|---|---|
| Two-Photon Fluorescence (2PF) | X | fs | 1 |
| Second Harmonic Generation (SHG) | fs | 1 | |
| Three-Photon Fluorescence (3PF) | X | fs | 1 |
| Third Harmonic Generation (THG) | fs | 1 | |
| Coherent anti-Stokes Raman Scattering (CARS) | ps or fs | 2 | |
| Stimulated Raman Scattering (SRS) | ps or fs | 2 | |
| Sum-Frequency Generation (SFG) | fs | 2 |
Table 1. Comparison of mechanisms used for nonlinear microscopy techniques.
Two-Photon Fluorescence Microscopy
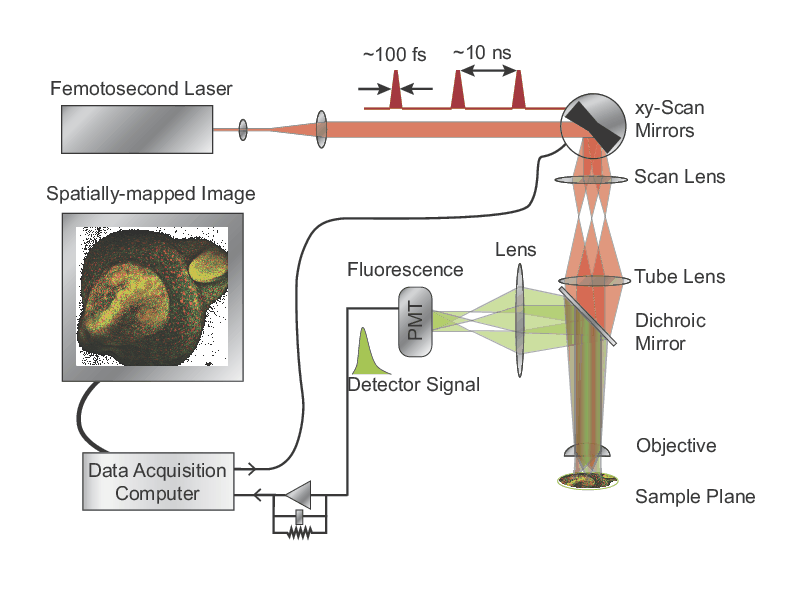
Three-Photon Fluorescence Microscopy

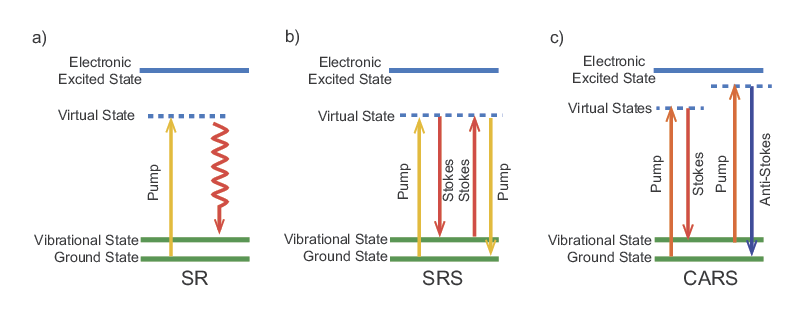
CARS and SRS Raman Microscopy
Bioimaging Products






For additional insights into photonics topics like this, download our free MKS Instruments Handbook: Principles & Applications in Photonics Technologies
Request a Handbook